
The following is a GUEST-POST composed by Dr. Sascha Tuchman, a friend and colleague from the University of North Carolina in Chapel Hill, NC. Dr Tuchman will be co-moderating the final pre-ASH 2018 #amyloidosis journal club (#amyloidosisJC) with Untangling Amyloidosis 208 Trainee Travel Grant Awardee Dr. Sam Rubinstein
 |
| Dr Tuchman |
 |
| Dr Rubinstein |
Background:
For basic information on transthyretin
(TTR) and how it forms amyloidosis, please refer to the background information
in recent blog post dated 10/28/18, which summarized the APOLLO study of patisiranin hereditary transthyretin polyneuropathy.
Drugs like patisian work to suppress TTR synthesis by the liver (“A” in
the below diagram). Another area of research interest has been in TTR stabilizers.
If we refer to the image below, we see that TTR circulates in blood as a
tetramer, meaning a single molecule made up of four pieces of TTR joined together.
The TTR tetramer dissociates, resulting in single TTR pieces (monomers). Those monomers can deposit in organs and form
amyloid. The theory behind TTR
stabilizers is that making it harder for the tetramers to dissociate means
there are fewer TTR monomers available to form amyloidosis (“B” in the below
diagram). The “B” approach was first successfully tested using the drug diflunisal
in patients with TTR neuropathy (Berk J et al., JAMA 2013). The current “ATTR-ACT” study examined the oral
TTR stabilizer tafamidis in patients with TTR amyloid cardiomyopathy.
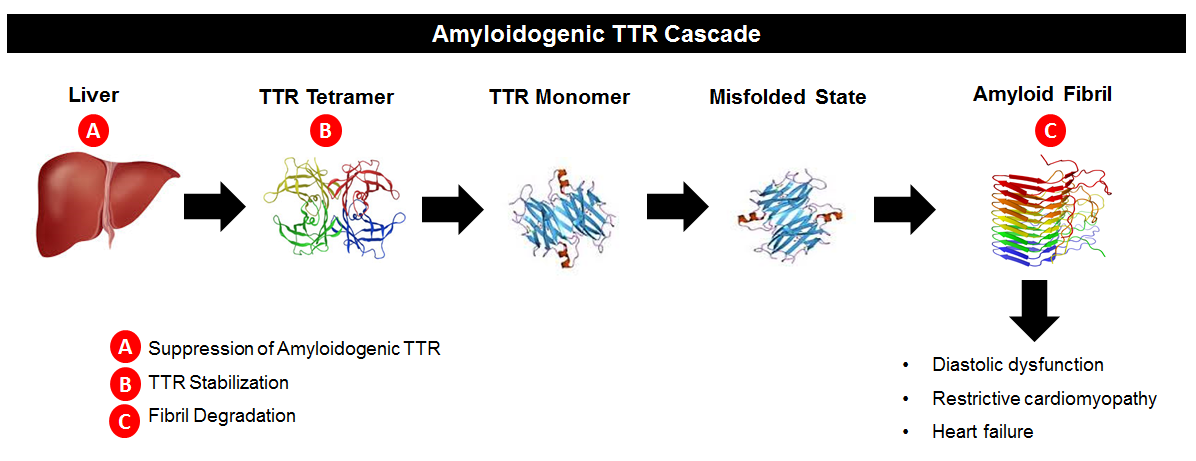
The above image is copied from: https://www.acc.org/latest-in-cardiology/articles/2015/10/13/08/35/emerging-therapies-for-transthyretin-cardiac-amyloidosis
Methods:
Phase 3, international study of 441 patients
with biopsy-proven TTR cardiac amyloidosis. Placebo-controlled and
double-blinded. Patients had to have
clinical evidence of active heart failure, but less severe than symptomatic at
rest (NYHA class <4). Severe
impairment of liver/kidney function or nutritional status precluded
participation. Patients were prohibited
from receiving other TTR-directed agents such as diflunisal or
doxycycline during study participation.
Patients received either 80 or 20 mg of tafamidis daily orally
or placebo (randomized in a 2:1:2 ratio). Stratified by TTR mutational status
(wild-type vs mutated) and baseline NYHA class. Treatment was for 30 months and
on completion, patients could opt to continue on to an extension study
including open label tafamidis.
The primary endpoint was all-cause mortality followed by
cardiovascular-related hospitalizations.
The planned sample size of 400 patients was 90% powered to detect a 30%
reduction in mortality and in cardiovascular-related hospitalizations from 2.5
to 1.5 over the 30 months on study.
Results:
548 patients screened and 441 randomized.
264 patients received tafamidis and 177 placebo. ~24% in both groups had
mutated TTR, with a similar frequency of specific TTR mutations in each group.
Other baseline factors were similar, including NYHA class, NT-proBNP, and body
mass index (BMI).
Both mortality and frequency of cardiovascular-related
hospitalizations were improved with tafamidis. Specifically the hazard ratio
for all-cause mortality was 0.7 (95% CI 0.51-0.96) and the relative risk for
hospitalizations was 0.68 (0.56-0.81).
The mortality benefit appeared after approximately 18 months on study. Benefits
were consistent across all subgroups except NYHA class 3 CHF, in which
mortality was the same but rate of hospitalizations was higher for tafamidis.
Functional measures of the heart also showed a slower
decline with tafamidis than placebo, such as distance walked in 6 minute walk
test and patient-reported symptoms.
There were no significant adverse events for tafamidis, and
discontinuation from study as a result of adverse events was more common for
placebo-treated than tafalidis-treated patients.
Conclusions:
Tafamidis reduced mortality and
cardiovascular-related hospitalization in patients with cardiomyopathy
resulting from either wild-type or mutated TTR.
Markers of heart function such as 6 minute walk test and NT-proBNP
suggest that tafamidis doesn’t stop the disease, rather it slows its
progression.
Comments:
Tafamidis is clearly a game-changer in the
sense that it’s the first medication that will likely be FDA-approved for TTR
cardiomyopathy. However, the fact that the disease still progresses indicates
that more needs to be done. In particular, combining tafamidis with drugs that
perturb other aspects of TTR formation could be promising. (Recall in the
diagram above that tafamidis is a “B” drug.
Other drugs such as patisiran and inotersen are “A” drugs. Combining a B drug with an A drug and/or a C
drug may improve the benefit overall to patients with these diseases by further
slowing or even reversing damage caused to the heart by TTR.)
EDITOR'S ADDENDUM: looking forward to this week's installment of #amyloidosisJC, at 8 pm EST on Sunday November 18, 2018. Remember to use the #amyloidosisJC hashtag when you log on! Also, hoping to see you in San Diego for the Untangling Amyloidosis 2018 Friday Satellite Symposium. Turns out Dr Tuchman is joining the faculty speaker roster!
